Medication Transformed
Oral liquid formulations of topiramate and zonisamide, the molecules you know and trust1-3
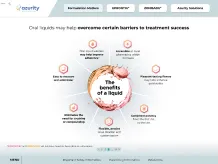
The FIRST ready-to-use liquid
topiramate approved by the US
Food & Drug Administration (FDA)
The ONLY ready-to-usea liquid zonisamide
The ONE and ONLY
alternative formulation to
solid oral doses of zonisamide
aShake well before administering.
Please note that ZONISADE® (zonisamide oral suspension) 100 mg/5 mL and EPRONTIA® (topiramate) oral solution, 25 mg/mL are 2 separate pharmaceutical products. ZONISADE® cannot be used as adjunctive therapy with EPRONTIA® or other forms of topiramate.
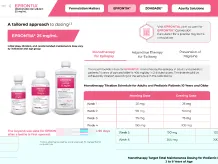
EPRONTIA® (topiramate) oral solution, 25 mg/mL
EPRONTIA is indicated for:
- Initial monotherapy for the treatment of partial-onset or primary generalized tonic-clonic seizures in patients 2 years of age and older.
- Adjunctive therapy for the treatment of partial-onset seizures, primary generalized tonic-clonic seizures, or seizures associated with Lennox-Gastaut syndrome in patients 2 years of age and older.
- Preventive treatment of migraine in patients 12 years of age and older.
Inform patients that a calibrated measuring device is recommended to accurately measure and deliver the prescribed dose. A household teaspoon or tablespoon is not an adequate measuring device.
Please see Important Safety Information throughout and the full Prescribing Information at EPRONTIA.com.
Important Safety Information
EPRONTIA® (topiramate) oral solution, 25 mg/mL
EPRONTIA is indicated for:
- Initial monotherapy for the treatment of partial-onset or primary generalized tonic-clonic seizures in patients 2 years of age and older.
- Adjunctive therapy for the treatment of partial-onset seizures, primary generalized tonic-clonic seizures, or seizures associated with Lennox-Gastaut syndrome in patients 2 years of age and older.
- Preventive treatment of migraine in patients 12 years of age and older.
Inform patients that a calibrated measuring device is recommended to accurately measure and deliver the prescribed dose. A household teaspoon or tablespoon is not an adequate measuring device.
Additional Important Safety Information
Warnings and Precautions:
Acute Myopia and Secondary Angle Closure Glaucoma Syndrome: A syndrome consisting of acute myopia associated with secondary angle closure glaucoma has been reported in patients receiving EPRONTIA (topiramate). Symptoms include acute onset of decreased visual acuity and/or ocular pain. Ophthalmologic findings can include some or all the following: myopia, mydriasis, anterior chamber shallowing, ocular hyperemia (redness), choroidal detachments, retinal pigment epithelial detachments, macular striae, and increased intraocular pressure. This syndrome may be associated with supraciliary effusion resulting in anterior displacement of the lens and iris, with secondary angle closure glaucoma. Symptoms typically occur within 1 month of initiating EPRONTIA therapy. The primary treatment to reverse symptoms is discontinuation of EPRONTIA.
Visual Field Defects: Visual field defects have been reported in clinical trials and in postmarketing experience in patients receiving topiramate. In clinical trials, most of these events were reversible after topiramate discontinuation. If visual problems occur, consideration should be given to discontinuing the drug.
Oligohidrosis and Hyperthermia: Oligohydrosis (decreased sweating), infrequently resulting in hospitalization, has been reported in association with EPRONTIA use. The majority of the reports have been in pediatric patients. Patients, especially pediatric patients, should be monitored closely for evidence of decreased sweating and increased body temperature, especially in hot weather. Caution should be used when EPRONTIA is prescribed with other drugs that predispose patients to heat-related disorders; these drugs include, but are not limited to, other carbonic anhydrase inhibitors and drugs with anticholinergic activity.
Metabolic Acidosis: Metabolic acidosis was commonly observed in adult and pediatric patients in clinical trials and is caused by renal bicarbonate loss due to carbonic anhydrase inhibition by EPRONTIA. Conditions or therapies that predispose patients to acidosis (such as renal disease, severe respiratory disorders, status epilepticus, diarrhea, ketogenic diet, or specific drugs) may be additive to the bicarbonate lowering effects of EPRONTIA. Topiramate treatment that causes metabolic acidosis during pregnancy can possibly produce adverse effects on the fetus and might also cause metabolic acidosis in the neonate from possible transfer of topiramate to the fetus. Baseline and periodic serum bicarbonate measurements are recommended during EPRONTIA treatment. If metabolic acidosis develops and persists, consideration should be given to either dose reduction or discontinuation of therapy using dose tapering.
Suicidal Behavior and Ideation: Antiepileptic drugs (AEDs), including EPRONTIA, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Cognitive/Neuropsychiatric Adverse Reactions: EPRONTIA can cause cognitive/neuropsychiatric adverse reactions. The most frequent of these can be classified into 3 categories: 1) cognitive-related dysfunction (eg, confusion, difficulty with concentration, difficulty with memory, speech or language problems), 2) psychiatric/behavioral disturbances (eg, depression or mood problems), and 3) somnolence or fatigue.
Fetal Toxicity: EPRONTIA can cause fetal harm when administered to pregnant women. Use during pregnancy can cause major congenital malformations, including but not limited to cleft lip and/or palate and being small for gestational age. The benefits and risks should be considered when administering this drug to women of childbearing potential.
Withdrawal of Antiepileptic Drugs: EPRONTIA should be gradually withdrawn to minimize the potential for seizures or increased seizure frequency. If rapid withdrawal is required, appropriate monitoring is recommended.
Decrease in Bone Mineral Density: EPRONTIA has been shown to decrease bone mineral density (BMD) and bone mineral content in pediatric patients. Decreased BMD was correlated with decreased serum bicarbonate, which commonly occurs with topiramate treatment and reflects metabolic acidosis, a known cause of increased bone resorption.
Negative Effects on Growth (Height and Weight): Negative effects on height and weight have been seen in pediatric patients receiving topiramate treatment. EPRONTIA may slow height increase and weight gain; carefully monitor children receiving prolonged topiramate therapy.
Serious Skin Reactions: Serious skin reactions (Stevens-Johnson Syndrome [SJS] and Toxic Epidermal Necrolysis [TEN]) have been reported in patients receiving topiramate. EPRONTIA should be discontinued at the first sign of a rash, unless the rash is clearly not drug related. If signs or symptoms suggest SJS/TEN, use of this drug should not be resumed, and alternative therapy should be considered. Inform patients about the signs of serious skin reactions.
Hyperammonemia and Encephalopathy (Without and With Concomitant Valproic Acid Use): EPRONTIA treatment can cause hyperammonemia with or without encephalopathy, the risk of which appears to be dose-related, and which has been reported more frequently with concomitant use of valproic acid. In patients who develop unexplained lethargy, vomiting, or changes in mental status associated with topiramate, hyperammonemic encephalopathy should be considered and an ammonia level should be measured.
Kidney Stones: EPRONTIA can cause an increased risk of kidney stones. The concomitant use of topiramate with any other drug producing metabolic acidosis, or potentially in patients on a ketogenic diet, may increase the risk of kidney stone formation. An increase in urinary calcium and a marked decrease in urinary citrate have been observed in topiramate-treated pediatric patients. This increased urinary calcium/citrate ratio increases the risk of kidney stones and/or nephrocalcinosis. Instruct patients to stay well hydrated while taking EPRONTIA.
Hypothermia With Concomitant Valproic Acid Use: Hypothermia has been reported in association with topiramate use with concomitant valproic acid both in conjunction with hyperammonemia and in the absence of hyperammonemia. Consider discontinuation of topiramate or valproate in patients who develop hypothermia. Clinical management should include examination of blood ammonia levels.
Adverse Reactions:
The most common adverse reactions associated with EPRONTIA include:
|
|
Use in Specific Populations
Pregnancy
Topiramate can cause fetal harm when administered to a pregnant woman. Small for gestational age (SGA) has been observed at all doses and appears to be dose dependent. Patients should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry collects information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll-free number 1-888-233-2334. Information about the North American Drug Pregnancy Registry can be found at http://www.aedpregnancyregistry.org/.
Lactation
Topiramate is excreted in human milk. The effects of topiramate on milk production are unknown. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for topiramate and any potential adverse effects on the breastfed infant from topiramate or the underlying maternal condition.
Women of Reproductive Potential
Women of childbearing potential who are not planning a pregnancy should use effective contraception because of the risks of oral clefts and SGA.
Renal Impairment
The clearance of topiramate is reduced in patients with moderate (creatinine clearance 30 to 69 mL/min/1.73 m2) and severe (creatinine clearance <30 mL/min/1.73 m2) renal impairment. A dosage adjustment is recommended in patients with moderate or severe renal impairment.
Patients Undergoing Hemodialysis
Topiramate is cleared by hemodialysis at a rate that is 4 to 6 times greater than in a normal individual. A dosage adjustment may be required.
The Important Safety Information does not include all the information needed to use EPRONTIA safely and effectively. Visit EPRONTIA.com for full Prescribing Information.
To report SUSPECTED ADVERSE REACTIONS, contact Azurity Pharmaceuticals, Inc., at 1-800-461-7449 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
© 2025 Azurity Pharmaceuticals, Inc.
PP-EPR-US-0416
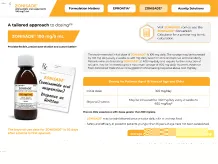
ZONISADE® (zonisamide oral suspension), 100 mg/5 mL
ZONISADE is indicated as adjunctive therapy for the treatment of partial-onset seizures in adults and pediatric patients 16 years of age and older.
Inform patients that a calibrated measuring device is recommended to measure and deliver the prescribed dose accurately. A household teaspoon or tablespoon is not an adequate measuring device.
Please see Important Safety Information throughout and the full Prescribing Information at ZONISADE.com.
Important Safety Information
ZONISADE® (zonisamide oral suspension), 100 mg/5 mL
ZONISADE is indicated as adjunctive therapy for the treatment of partial-onset seizures in adults and pediatric patients 16 years of age and older.
Inform patients that a calibrated measuring device is recommended to measure and deliver the prescribed dose accurately. A household teaspoon or tablespoon is not an adequate measuring device.
ADDITIONAL IMPORTANT SAFETY INFORMATION
Contraindications
Known hypersensitivity to sulfonamides, zonisamide, or any formulation ingredients.
Warnings and Precautions
Potentially fatal reaction to Sulfonamides: Fatalities have occurred as a result of severe reactions to sulfonamides (ZONISADE is a sulfonamide) including Stevens-Johnson syndrome, toxic epidermal necrolysis, fulminant hepatic necrosis, agranulocytosis, aplastic anemia, and other blood dyscrasias. If signs of hypersensitivity or other serious reactions occur, discontinue ZONISADE immediately.
Serious Skin Reaction: Serious skin reactions (Stevens-Johnson syndrome and toxic epidermal necrolysis) have been reported. Consideration should be given to discontinuing ZONISADE in patients who develop an otherwise unexplained rash. If the drug is not discontinued, patients should be observed frequently. Inform patients about the signs of serious skin reactions.
Serious Hematologic Events: Aplastic anemia and agranulocytosis have been reported in patients who received zonisamide treatment. There is inadequate information to assess the relationship, if any, between dose and duration of treatment and these events.
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity: Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multi-organ hypersensitivity, has occurred with zonisamide. Some of these events have been fatal or life-threatening. If signs or symptoms of DRESS are present, the patient should be evaluated immediately. ZONISADE should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
Oligohidrosis and Hyperthermia in Pediatric Patients: ZONISADE is not approved for use in pediatric patients below 16 years of age. Pediatric patients appear to be at an increased risk for zonisamide-associated oligohidrosis and hyperthermia. Patients, especially pediatric patients, treated with ZONISADE should be monitored closely for evidence of decreased sweating and increased body temperature, especially in warm or hot weather. Caution should be used when ZONISADE is prescribed with other drugs that predispose patients to heat-related disorders; these drugs include but are not limited to, carbonic anhydrase inhibitors and drugs with anticholinergic activity.
Acute Myopia and Secondary Angle Closure Glaucoma: Acute myopia and secondary angle closure glaucoma have been reported in patients receiving zonisamide. Elevated intraocular pressure can lead to serious sequelae, including permanent vision loss if left untreated. Symptoms typically occur within one month after initiating zonisamide therapy. The primary treatment to reverse symptoms is the discontinuation of zonisamide. Myopia and secondary angle closure glaucoma usually resolve or improve after discontinuation of zonisamide.
Suicidal Behavior and Ideation: Antiepileptic drugs (AEDs), including ZONISADE, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Metabolic Acidosis: Metabolic acidosis was commonly observed in adults and pediatric patients in clinical trials and is caused by renal bicarbonate loss due to the inhibitory effect of zonisamide on carbonic anhydrase. Pediatric patients may be more likely to develop metabolic acidosis than adults. Conditions or therapies that predispose to acidosis (such as renal disease, severe respiratory disorders, status epilepticus, diarrhea, ketogenic diet, or specific drugs) may be additive to the bicarbonate lowering effects of zonisamide. Measurement of baseline and periodic serum bicarbonate during treatment is recommended. If metabolic acidosis develops, consideration should be given to either dose reduction or discontinuation of therapy using dose tapering.
Seizures on Withdrawal: As with other AEDs, abrupt withdrawal of ZONISADE in patients with epilepsy may precipitate increased seizure frequency or status epilepticus. Dose reduction or discontinuation of ZONISADE should be done gradually.
Teratogenicity: ZONISADE may cause fetal harm when administered to pregnant women and should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Women of childbearing potential who are given ZONISADE should be advised to use effective contraception.
Cognitive/Neuropsychiatric Adverse Reactions: Use of zonisamide was frequently associated with central nervous system-related adverse reactions including psychiatric symptoms, cognitive dysfunction, and somnolence or fatigue.
Hyperammonemia and Encephalopathy: Hyperammonemia and encephalopathy have been reported with the postmarketing use of zonisamide. The risks may be increased in patients treated with zonisamide and concomitantly taking valproic acid or topiramate. Measure serum ammonia concentration if signs or symptoms of encephalopathy occur. Hyperammonemia resulting from zonisamide resolves when zonisamide is discontinued. Hyperammonemia from zonisamide may resolve or decrease in severity with a decrease of the daily dose.
Kidney Stones: ZONISADE may increase the risk for kidney stones. Instruct patients to stay well hydrated while taking ZONISADE.
Effect on Renal Function: ZONISADE can cause an increase in serum creatinine and blood urea nitrogen (BUN). ZONISADE should be discontinued in patients who develop acute renal failure or a clinically significant sustained increase in the creatinine/BUN concentration. ZONISADE should not be used in patients with renal failure (estimated [GFR] < 50 mL/min) as there has been insufficient experience concerning drug dosing and toxicity.
Status Epilepticus: Status epilepticus has been reported at a rate of 1% across all controlled and uncontrolled epilepsy studies in patients treated with zonisamide.
Laboratory Tests: ZONISADE may increase serum chloride and alkaline phosphatase, and decrease serum bicarbonate, phosphorus, calcium, and albumin. Clinical management should include a periodic assessment of laboratory values.
Adverse Reactions
The most common adverse reactions with ZONISADE (an incidence at least 4% greater than placebo) in controlled clinical trials and shown in descending order of frequency were somnolence, anorexia, dizziness, ataxia, agitation/irritability, and difficulty with memory and/or concentration.
Drug Interactions
ZONISADE should be used with caution if used in combination with alcohol or other CNS depressants.
Concomitant use of ZONISADE with any other carbonic anhydrase inhibitor may increase the severity of metabolic acidosis and may also increase the risk of kidney stone formation.
USE IN SPECIFIC POPULATIONS
Pregnancy
ZONISADE may cause serious adverse fetal effects, based on clinical and nonclinical data.
Advise pregnant patients to enroll in the NAAED Pregnancy Registry. This can be done by calling the toll-free number 1-888-233-2334 and must be done by patients themselves.
Lactation
Zonisamide is excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from ZONISADE, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Females and Males of Reproductive Potential
Based on animal data, ZONISADE can cause fetal harm when administered to a pregnant woman, and female fertility may be compromised with ZONISADE.
Pediatric Use
The safety and effectiveness of ZONISADE in pediatric patients below the age of 16 have not been established.
The Important Safety Information does not include all the information needed to use ZONISADE safely and effectively. For additional safety information, please see the accompanying full Prescribing Information for ZONISADE.
To report SUSPECTED ADVERSE REACTIONS, contact Azurity Pharmaceuticals, Inc. at 1-800-461-7449 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
© 2025 Azurity Pharmaceuticals, Inc.
PP-ZON-US-0060